Software
The following software tools and services, developed by MMBioS partners, are freely available to the biomedical community. We ask that any publications resulting from their use include an acknowledgement.

AlignTK
AlignTK is an image alignment toolkit. It is designed for batch-oriented alignment of a large number of 2-D images in either 2 or 3-dimensions. Although it has been applied most extensively to electron-microscopy (EM) images of neural tissue, the package can be used with arbitrary grayscale images. Multiple image formats are supported.
| More : | >> Home page | >> Downloads |
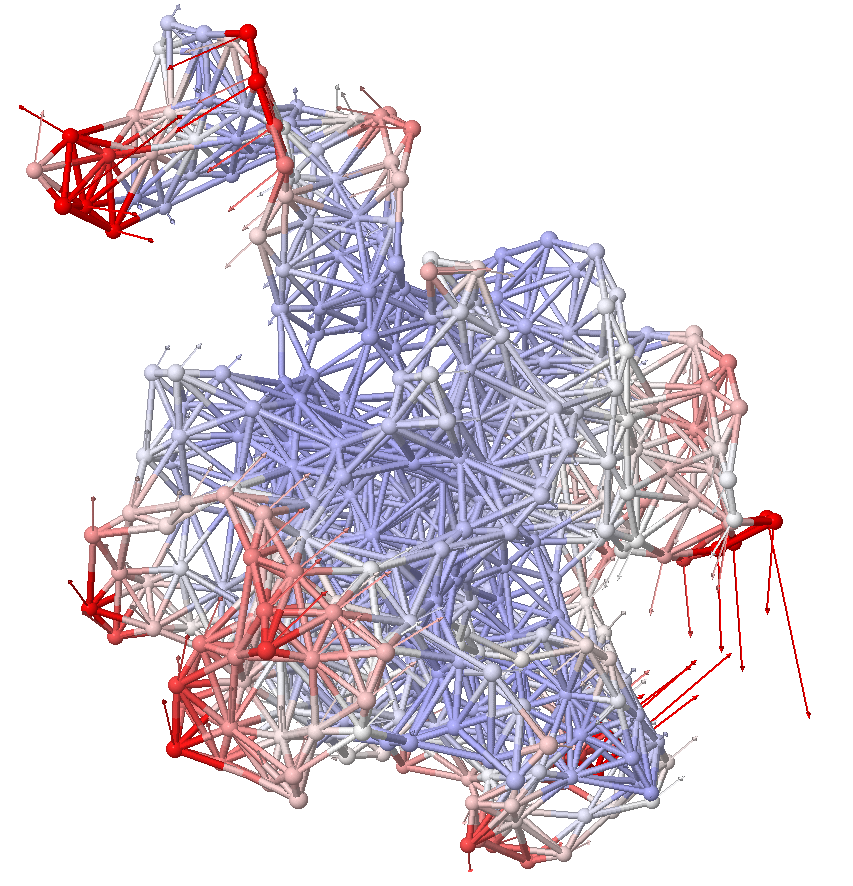
ANM
ANM (Anisotropic network model) t is a simple tool for predicting the collective motions of molecular systems introduced using Elastic Network Models (ENM) and normal mode analysis. The biomolecular system is represented as a network, or graph, the nodes of which are the residues and the springs are their interactions. The model helps elucidate the intrinsic dynamics of proteins and their complexes and make inferences on functional mechanisms.
| More : | >> Home page |

BioNetGen
BioNetGen is software for the specification and simulation of rule-based models of biochemical systems, including signal transduction, metabolic, and genetic regulatory networks.
| More : | >> Home page |
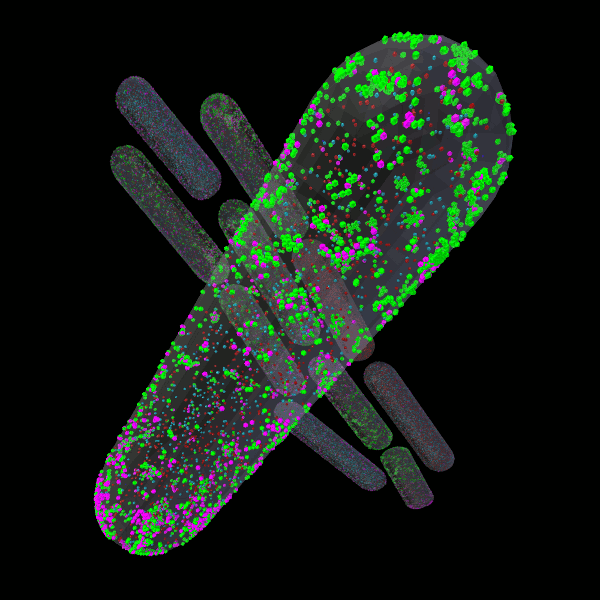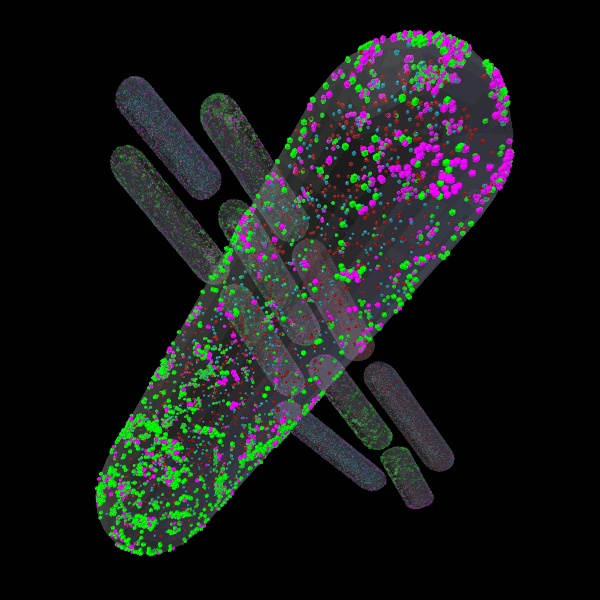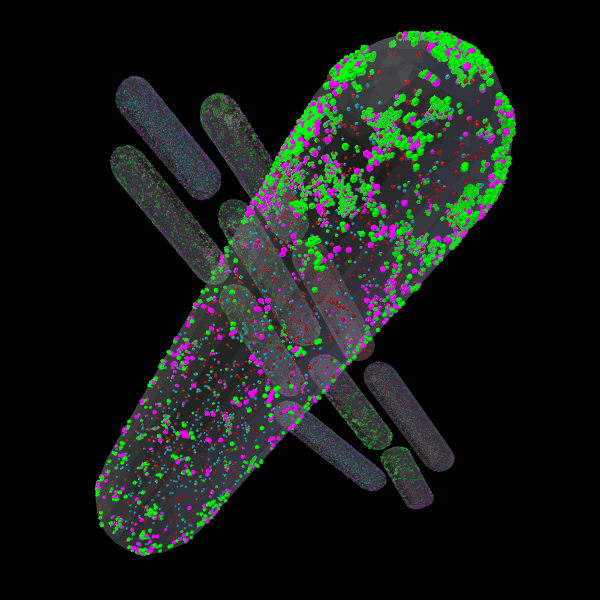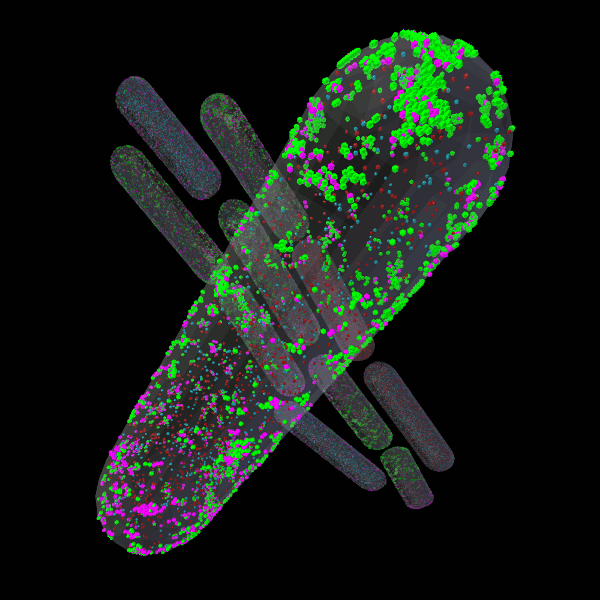
CellBlender
CellBlender is a Blender addon for creation, simulation, visualization, and analysis of realistic 3D Cell Models. CellBlender leverages the full-featured 3D content creation capabilities of Blender to support a rich environment for the creation of simulation-ready, biophysically realistic models of the microscopic structure and biochemical function of cells.
CellBlender is fully functional with MCell and partially functional with SBML (http://sbml.org). We invite the computational cell biology community to contribute to the project, adding features and support for their favorite simulation environments.
| More: | >>Home page | >>Downloads | >>Forum |

CellOrganizer
The CellOrganizer project provides tools for
- learning generative models of cell organization directly from images
- storing and retrieving those models in XML files
- synthesizing cell images (or other representations) from one or more models
Model learning captures variation among cells in a collection of images. Images used for model learning and instances synthesized from models can be two- or three-dimensional static images or movies.
| More: | >> Home page | >> Tutorials |
GAMELAN
GamelanPy is a Python implementation of the GAMELAN (GrAphical Models of Energy LANdscapes) algorithm. This algorithm is for learning a sparse Gaussian Mixture Model for structural fluctuations of a protein, but can also be used for other data where the original data can be assumed to follow a mixture of Gaussian distributions. GamelanPy supports options for sub-sampling methods for scalability and nonparanormal distributions for richer family of distributions than Gaussians. This package contains a pure python library and scripts for the command-line usages.
More information on GAMELAN can be found in this paper: Generative Models of Conformational Dynamics
| More : | >> Home page |
iGNM
Gaussian network model (GNM) is a simple yet powerful model for investigating the dynamics of proteins and their complexes. GNM analysis became a broadly used method for assessing the conformational dynamics of biomolecular structures with the development of a user-friendly interface and database, iGNM, in 2005. We present here an updated version, iGNM 2.0 http://gnmdb.csb.pitt.edu/, which covers more than 95% of the structures currently available in the Protein Data Bank (PDB). Advanced search and visualization capabilities, both 2D and 3D, permit users to retrieve information on inter-residue and inter-domain cross-correlations, cooperative modes of motion, the location of hinge sites and energy localization spots. The ability of iGNM 2.0 to provide structural dynamics data on the large majority of PDB structures and, in particular, on their biological assemblies makes it a useful resource for establishing the bridge between structure, dynamics and function.
| More : | >> Home page | >> Tutorials |
GTKDynamo
GTKDynamo is free/open source software which, together with pDynamo, transforms PyMOL into a powerful interface for molecular modeling. The interface has been designed to facilitate determining reaction pathways in biological systems, specially using hybrid QC/MM (or QM/MM) methods. Pymol has been chosen as a graphical interface to pDynamo because it has a python API with wide documentation available.
| More : | >> Home page | >> Tutorials | >> Downloads |
MCell
MCell is a Monte Carlo reaction-diffusion simulator for modeling computational microphysiology in arbitrarily complex 3D spatial geometries.
As a modeling tool, MCell creates realistic simulations of cellular signaling in the complex 3-D subcellular microenvironment in and around living cells -- what we call cellular microphysiology. At such small subcellular scales the familiar macroscopic concept of concentration is not useful and stochastic behavior dominates. MCell uses highly optimized Monte Carlo algorithms to track the stochastic behavior of discrete molecules in space and time as they diffuse and interact with other discrete effector molecules (e.g. ion channels, enzymes, transporters) heterogeneously distributed within the 3-D geometry of the subcellular environment.
| More: | >> Home page | >> Documentation | >> Tutorials | >> Downloads |
| >> Forum |
ProDy
ProDy is a free and open-source Python package for protein structural dynamics and sequence evolution analysis. It is designed as a flexible and responsive API suitable for interactive usage and application development. NMWiz, a VMD plugin GUI, also accompanies ProDy for streamlining protein dynamics analysis calculations and enabling comparative visual analysis of experimental and theoretical data.
With ProDy, you can perform principal component analysis of heterogeneous X-ray structures, NMR models, and MD snapshots. Protein dynamics can be modeled using normal mode analysis of anisotropic network model with optional distance and property dependent force constants. Powerful and customizable atom selections allow for contact identification and matching, superposing, and comparing multiple structures/chains. Newest additions to ProDy include fast and flexible features for analysis of sequence evolution and its comparison to protein functional dynamics.
| More: | >> Home page | >> Tutorials | >> Downloads |
Rhapsody
Rhapsody is a web tool for pathogenicity prediction of human missense variants based on sequence, structure and dynamics of proteins.
| More: | >> Home page |
SWiFT-IR
SWiFT-IR uses Signal Whitening Fourier Transform Image Registration technique to achieve high precision image matching which is very robust to typical image distortions and defects. This high quality image matching in turn allows a hierarchical model based approach for the deformable registration of very deep image serial-section electron microscopy datasets. SWiFT-IR is also useful with other forms of grayscale and color imagery.
| More: | >> Home page |

WE
The "weighted-ensemblizer" is an automated tool for setting up WESTPA-based weighted-ensemble (WE) simulations for existing MCell models. Because MCell models can be complex and expensive to simulate, WE can provide enhanced sampling for targeted observables of interest, such as the concentration of a certain species in a specified location, by using a strategy of replicating and pruning trajectories. The alpha-version webserver was developed by Rory Donovan, a student in the Zuckerman research group supported by the MMBioS resource.
For more information, see: Donovan RM, Tapia JJ, Sullivan DP, Faeder JR, Murphy RF, Dittrich M, Zuckerman DM. (2016) Unbiased Rare Event Sampling in Spatial Stochastic Systems Biology Models Using a Weighted Ensemble of Trajectories. PLoS Comput Biol 12(2): e1004611. doi: 10.1371/journal.pcbi.1004611
WESTPA
WESTPA (the Weighted Ensemble Simulation Toolkit with Parallelization and Analysis) is an open-source software package that provides a high-performance framework for carrying out extended-timescale simulations of rare events with rigorous kinetics using the weighted ensemble algorithm of Huber and Kim (1996). The software also includes options for further enhancing the sampling efficiency through reassignment of weights according to either equilibrium or nonequilibrium steady state, and a plugin for using a weighted ensemble-based string method. The software is designed to interface with any stochastic simulation engine, including but not limited to molecular dynamics (e.g. AMBER, GROMACS, and NAMD), Monte Carlo codes, BioNetGen, and MCell.
| More: | >> Home page |